
A research group led by Professor Hideaki Matsui, Professor Akiyoshi Kakita, Professor Osamu Onodera, and others at the Brain Research Institute, Niigata University, published a Parkinson's disease model on May 25 revealing that mitochondrial DNA leakage into the cytoplasm causes inflammatory responses, cell death, and neurodegeneration. The research group confirmed mitochondrial DNA leakage into the cytoplasm using various Parkinson's disease models, such as cultured cells and zebrafish. Manipulations of these models, such as accelerated degradation, were found to improve the inflammatory response and neurodegeneration. Similar phenomena were observed in human brains measured during autopsies. This outcome is expected to lead to the development of therapy for patients with Parkinson's disease and the results have been published in the British scientific journal Nature Communications.
Parkinson's disease is an intractable neurological disease (unknown origin and untreatable) characterized by progressive motor symptoms caused by a decrease of dopamine-producing neurons in the substantia nigra. Onset is most common in individuals over the age of 50, but juvenile cases also exist. Much remains unknown about the disease state. The involvement of mitochondrial and lysosomal dysfunction has been reported; however, the detailed mechanisms of this involvement are not understood. Previously, this research group discovered that the African killifish, a short-lived fish species that is alive only during the rainy season in Mozambique, Africa, displays a phenotype similar to Parkinson's disease as they age. This observation revealed the leakage of mitochondrial DNA into the cytoplasm.
In this study, by examining cultured Parkinson's disease cells and zebrafish, researchers clarified the occurrence of a similar mitochondrial DNA leakage. In addition, it was confirmed that the leaked mitochondrial DNA induced cytotoxicity and neurodegeneration in cultured cells and zebrafish. In cultured cells, decreased PINK1, GBAs, and ATP13A2 levels, which are related to Parkinsonian diseases, increased cytosolic mitochondrial-derived DNA and induced type 1 interferon responses and cell death. In healthy individuals, even if mitochondrial damage occurs and DNA leaks into the cytosol, degradation by deoxyribonuclease II (DNaseII) and other enzymes present in lysosomes occurs. Therefore, it was hypothesized that the decomposition by DNaseII is not maintained in Parkinson's disease. However, because DNA sensors such as IFI16 continue to work, inflammatory responses and cell death are triggered, and verification was performed. Specifically, the researchers created a system in which DNaseII was overexpressed in the cultured Parkinson's disease cells and used RNA-interference and genome-editing methods to suppress the function of IFI16 They then examined the effects of these manipulations on cell death. They found that in both systems cell death was improved.
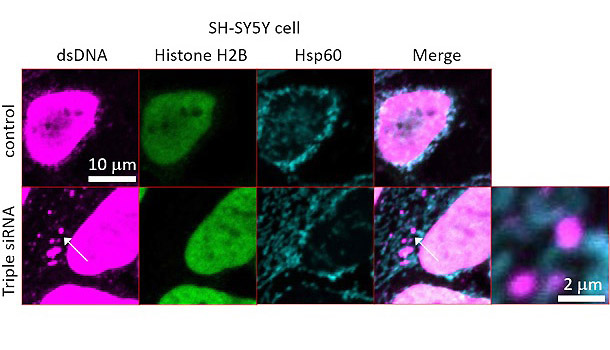
Credit: Matsui et al., Nat Commun. 2021
Through these manipulations the researchers confirmed that even in zebrafish, overexpressing human DNaseII in the GBA-mutant Parkinson's disease model improved motor deficits and the degeneration of dopaminergic neurons. Cytoplasmic DNA from IFI16 and mitochondria were confirmed to accumulate at lesion sites obtained from pathological autopsies of individuals with Parkinson's disease. "We were able to find mitochondrial DNA that leaked into the cytoplasm, which is central to neuronal cell death," Dr. Matsui said. "We would like to continue research that links knowledge to therapeutic outcomes in the future."
This article has been translated by JST with permission from The Science News Ltd.(https://sci-news.co.jp/). Unauthorized reproduction of the article and photographs is prohibited.