
A research group consisting of Professor Motoi Kanagawa of the Graduate School of Medicine, Ehime University, Professor Tatsushi Toda of the Graduate School of Medicine, the University of Tokyo, Doctor Hideki Tokuoka of the Graduate School of Medicine, Kobe University, and their colleagues announced that, working with Tokyo Metropolitan Geriatric Hospital and Institute of Gerontology, Nippon Medical School Foundation and Radboud University (the Netherlands), they have succeeded in creating and treating a mouse model for muscular dystrophy due to glycosylation defects. They confirmed that it is possible to treat this disease through the supplementation of glycan materials that increased delivery efficacy within cells in the mouse model and ascertained that gene therapies can inhibit the progress of the disease. It is hoped that their results will lead to the development of treatments for a wide range of diseases caused by glycosylation defects. The outcomes of the study were published in the April 14 edition of Nature Communications, an international science journal.
Provided by Motoi Kanagawa, Ehime University and modified by JST
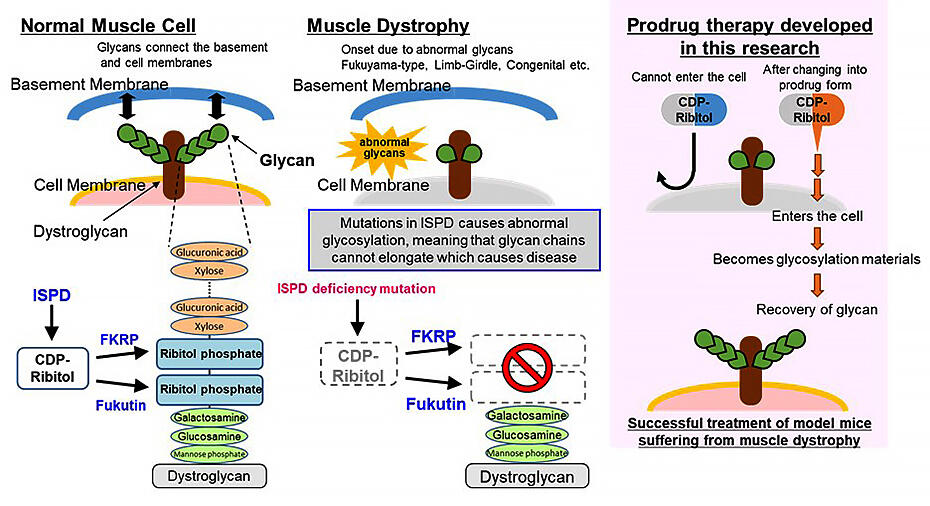
Muscular dystrophy is the general term for hereditary diseases that cause muscular degeneration and necrosis. There are several types, with different causative genes and severity. Of these, Fukuyama congenital muscular dystrophy (FCMD) is an autosomal recessive hereditary disease specifically prevalent among Japanese people; 1 in 90 Japanese people carry the pathogenic factors for this disease, and it has the second highest prevalence rate among infant muscular dystrophies. In addition to a progressive decline of muscle strength, it can be accompanied by gyrus and eye abnormalities. Many patients die before they reach 20 years of age, and there is no treatment.
So far, the group has clarified the causative gene of FCMD (fukutin) in 1998 and discovered that glycosylation defects occur with this disease in 2001. In 2009, it was elucidated that muscle cell necrosis in muscular dystrophy due to glycosylation defects occurs because the bond between the dystroglycan (a glycoprotein) on the surface of the muscle cells and the basal membrane degrades as a result of glycosylation defects. In 2016, they clarified that a novel compound (ribitol phosphate) is contained in the glycans on the muscle cell surface; the enzyme group associated with its biosynthesis, CDP-ribitol, which is a ribitol phosphate material, and its synthesis enzyme (isoprenoid synthase domain-containing protein (ISPD)). Since these reports, researchers have learned that abnormalities in glycans, which bond to proteins and lipids and are responsible for diverse physiological functions in the body, are associated with many diseases.
In this research, the group produced muscle-selective ISPD-deficient mice with the aim of creating a disease mouse model for research into treatments, based on past research. When they did so, they replicated muscular dystrophy due to glycosylation defects. The muscle strength and body weight of the mice decreased, the CDP ribitol concentration in the muscles decreased, there were glycosylation defects, and the mice died by 20 weeks of age. Next, to investigate whether treatments had an effect after the onset of the disease, the group carried out gene therapy that used adeno-associated virus (AAV) vectors to deliver ISPD genes to four-week-old mice. The results indicated that this inhibits the progress of the disease, and that treatment is possible. It also demonstrated the feasibility of CDP-ribitol replacement therapy. On the other hand, it was thought that CDP-ribitol has poor delivery efficacy within cells and low stability within the body, even if it is administered from outside the cells.
Thus, the group investigated the creation of prodrugs that improve deliverability through chemical modification and are metabolically converted to bioactive CDP-ribitol in the cells. More specifically, they explored different types and locations of functional groups, and prepared 10 different compounds. These were added to the cells of the ISPD-deficient mice, and the drug efficacy was evaluated using glycosylation recovery and toxicity as indicators.
The results showed that tetraacetylated CDP-ribitol (TetA) with four acetyl group bonds was the most effective of these drugs. Thus, the group administered TetA to the mouse model over a longer period of time and observed its therapeutic efficacy. They were able to confirm partial glycosylation recovery and a clear improvement in the mice's pathological condition when TetA was administered twice a week from when the mice were four weeks old to seven weeks old. Moreover, when TetA was added to the cells of two patients with different diseases (Walker-Warburg syndrome phenotype and muscle-eye-brain disease phenotype), glycosylation recovery was recognized in both cases, indicating the possibility that this will be effective in humans too. It is possible that gene therapy and prodrug therapy based on these outcomes can be applied to other patients with mutations in enzymes that use CDP-ribitol as a substrate, including Fukuyama congenital muscular dystrophy.
The research group is currently aiming to create prodrugs with higher delivery efficacy and searching for corporate partners to carry out joint research.
This article has been translated by JST with permission from The Science News Ltd.(https://sci-news.co.jp/). Unauthorized reproduction of the article and photographs is prohibited.