
The research group led by Project Associate Professor Satoru Torii and Professor Shigeomi Shimizu of the Medical Research Institute (MRi) at Tokyo Medical and Dental University (TMDU), in collaboration with the group led by Associate Professor Shigeto Sato and Professor Nobutaka Hattori of the Graduate School of Medicine at Juntendo University, announced that they have clarified that Parkinson's disease is caused by PARK22 /CHCHD2 gene mutations. The result was published in the online edition of EMBO Molecular Medicine.
The onset of Parkinson's disease is known to be associated with the degeneration and loss of dopaminergic neurons in the substantia nigra of the midbrain. Most cases of Parkinson's disease are sporadic (meaning they occur in people with no known family history of the disorder). However, familial Parkinson's disease (i.e., there is a history of family members with the disorder) is caused by mutations in genes such as the one coding for α-synuclein.
Although CHCHD2 has recently been revealed as the causative gene for familial Parkinson's disease (PARK22) as well as a risk gene for sporadic Parkinson's disease, the mechanism underlying the onset of Parkinson's disease remains unclear, and many of its aspects and abnormalities are unknown.
The research group clarified that the pathogenic mechanism of Parkinson's disease is caused by the most common CHCHD2 mutation T61I, which results in the 61st threonine being replaced by isoleucine in the protein.
When mouse-derived neuroblastoma Neuro2a cells differentiate into neurons, the CHCHD2 wild-type (CHCHD2WT) remains localized in the mitochondria, whereas the T61I mutant (CHCHD2T61I) enters the mitochondria once and then leaves the organelle to move into the cytosol. After recruiting casein kinase I isoform epsilon/delta (CK1ε/δ), the mislocalized CHCHD2T61I induces phosphorylation of both α-synuclein and neurofilament light chain (NEFL), and these molecules then form aggresomes.
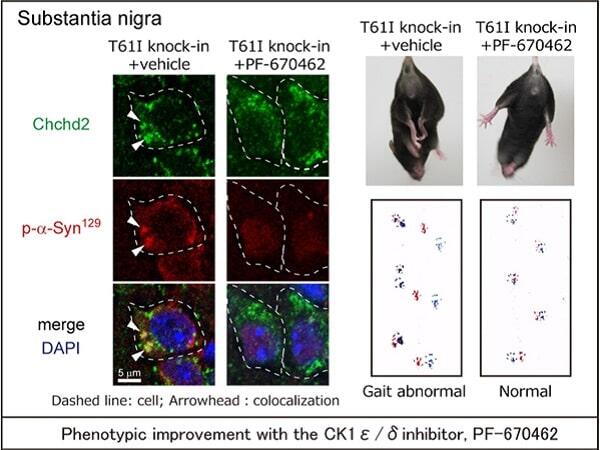
The analysis with mice showed that aggresomes containing CHCHD2T61I, CK1ε/δ, phosphorylated α-synuclein, and phosphorylated NEFL were formed in dopaminergic neurons in both CHCHD2T61I knock-in mice and transgenic mice. The animals in these two groups exhibited locomotion abnormalities and motor dysfunction characteristic of neurodegeneration. Similar aggresomes were found in the postmortem brain of patients with Parkinson's disease and in dopaminergic neurons generated from patient-derived induced pluripotent stem (iPS) cells.
When the cells and mice were treated with PF670462 (a CK1ε/δ inhibitor), the phosphorylation of α-synuclein and NEFL was significantly suppressed. PF670462 also suppressed the death of dopaminergic neurons and improved neurodegenerative phenotypes, such as clasping and gait abnormalities, in the CHCHD2T61I knock-in mutant mice.
Torii said, "We are conducting experiments focusing on the T61I mutation of CHCHD2. We believe that the phenomenon may be associated with sporadic Parkinson's disease, the cause of which is still unknown. Since the phosphorylation of α-synuclein is the common feature, we would like to target this step to develop a treatment for sporadic Parkinson's disease."
Journal Information
Publication: EMBO Molecular Medicine
Title: Involvement of casein kinase 1 epsilon/delta (Csnk1e/d) in the pathogenesis of familial Parkinson's disease caused by CHCHD2
DOI: 10.15252/emmm.202317451
This article has been translated by JST with permission from The Science News Ltd. (https://sci-news.co.jp/). Unauthorized reproduction of the article and photographs is prohibited.