
A research group led by Lecturer Kotaro Tsuboyama of the Institute of Industrial Science (IIS) at the University of Tokyo (a postdoctoral fellow at Northwestern University at the time of the research) and Assistant Professor Gabriel J. Rocklin of Northwestern University has developed an efficient method for measuring the folding stability of proteins. The folding stability of a protein is an indicator of how easily it maintains a specific functional conformation, and it defines the percentage of molecules that exhibit a certain function. Until recently, the stability of only one type of protein could be measured in a single experiment. However, with the new method, the stability of up to 900,000 types of proteins can be measured in a single experiment. The results have been published in Nature.
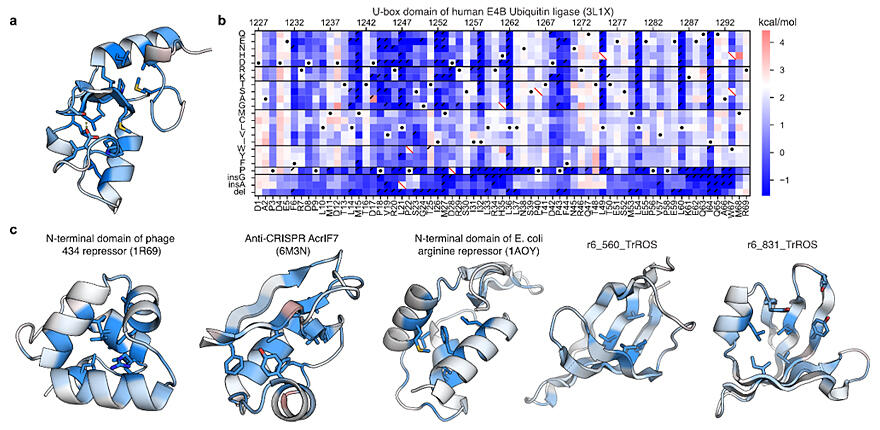
(a,c) The importance of each protein position to folding stability. (more important in blue, less important in red).
(b) Original data for the structural diagram of the protein in (a). It shows that the protein is structurally less stable (blue) or more stable (red) than that of the wild-type protein (white and black dots) To create a diagram of one protein, the folding stability of more than a thousand different amino acid sequences must be measured. The horizontal axis indicates position in the protein, and the vertical axis indicates amino acid substitutions, deletions, or insertions.
Provided by the University of Tokyo
Anfinsen's dogma is well known in molecular biology. It states that for proteins in living organisms, "The amino acid sequence determines the structure of the protein and further determines its function." In almost all proteins the structure determines function. Additionally, most proteins switch between multiple states, including the unstructured unfolded and properly folded conformational states. Therefore, 'protein folding stability,' which indicates the percentage of molecules with a properly folded structure, is one of the most important amounts of characteristics of a protein since it indicates the percentage of functional proteins.
A decrease in protein folding stability can lead to various diseases, such as cancer, due to unintended interactions between proteins or a lack of function. Despite this importance, it has been difficult to gain an accurate understanding and predict the stability of proteins from their amino acid sequences and structures. This is because the experiments involved are too labor-intensive, costly, and time-consuming. Even the database with the most entries of protein folding stability measurements made over the past several decades contains information on only around 30,000 proteins. On top of this, the data uploaded to the database are from different published studies in which the measurement conditions and data quality varied, making it difficult to understand protein folding stability comprehensively.
In this study, the research group has realized an efficient method for measuring the folding stability of proteins by developing two major techniques. The first is to convert the amino acid sequence information of the protein into DNA nucleic acid information. The amino acid sequence can be identified by reading the cDNA sequence using the cDNA display method, linking each protein to its corresponding cDNA. Combining two techniques (i.e., cDNA display method and next-generation DNA sequencing) makes it possible to analyze the amino acid sequences of a huge number of different proteins at once.
The second technique involves using the protein-cleaving enzyme protease to quantify the stability of the protein. Because proteases can degrade proteins in an unstable and unwound state, they cleave proteins with high stability slowly and those with low stability quickly. In other words, measuring the rate at which a protein cleaves makes it possible to measure its stability.
These two techniques allowed the research group to measure the folding stability of approximately 900,000 different proteins in a single experiment. These experiments were repeated several times, and high-quality data were selected to create a publicly available database of the stability of approximately 800,000 proteins. In addition to its size, its other major advantage is that the measurement conditions used to obtain the data are identical.
The mechanisms that maintain the structure of each protein have been studied in approximately 500 natural and artificial proteins. For each protein, stability was measured after replacing one amino acid at each position with another of 20 different amino acids, deleting each amino acid, and inserting glycine or alanine. This allowed the researchers to visualize the stability of each protein, such as which sites are important for structural maintenance.
The relationship between the ease with which each amino acid is utilized in proteins in the organism and the structural stability that the amino acid imparts was also studied. More structure-stabilizing amino acids tended to be used more often, and this trend could be quantified. Furthermore, the research group studied how amino acids are used when the effect of folding stability is eliminated. The results showed that hydrophilic amino acids, which are more soluble in water, are more likely to be used. By contrast, the less water-soluble hydrophobic amino acids, especially aromatic amino acids containing aromatic rings, tend to be used less often.
The sheer size of this database, which is much larger than other existing databases, has made it possible to infer important properties that define the stability of proteins and propose general rules related to folding stability. Large folding stability datasets like this also provide the basis for AI development in protein science. For example, such AI is expected to aid in the identification of disease-causing amino acid mutations and facilitate the more efficient synthesis of protein drugs.
Journal Information
Publication: Nature
Title: Mega-scale experimental analysis of protein folding stability in biology and design
DOI: 10.1038/s41586-023-06328-6
This article has been translated by JST with permission from The Science News Ltd. (https://sci-news.co.jp/). Unauthorized reproduction of the article and photographs is prohibited.