
An international collaborative group led by Team Leader Takaomi Saido of the RIKEN Center for Brain Science and Professor Nobuhisa Iwata of Nagasaki University's Graduate School of Biomedical Sciences has announced that they elucidated the mechanism by which amyloid beta (Aβ) is transformed into its pathological form that is resistant to break down in the Alzheimer's disease (AD) brain. The group analyzed Alzheimer's model mice manipulated so that the Aβ-degrading enzyme neprilysin does not work. In these mice, enzymes different from those working in the normal brain degraded Aβ, and this process was shown to involve a modification imparting resistance to degradation, leading to the accumulation of pathological Aβ. The findings are expected to contribute to the development of new therapeutic agents and were published in the international journal Life Science Alliance on September 30.
Provided by RIKEN
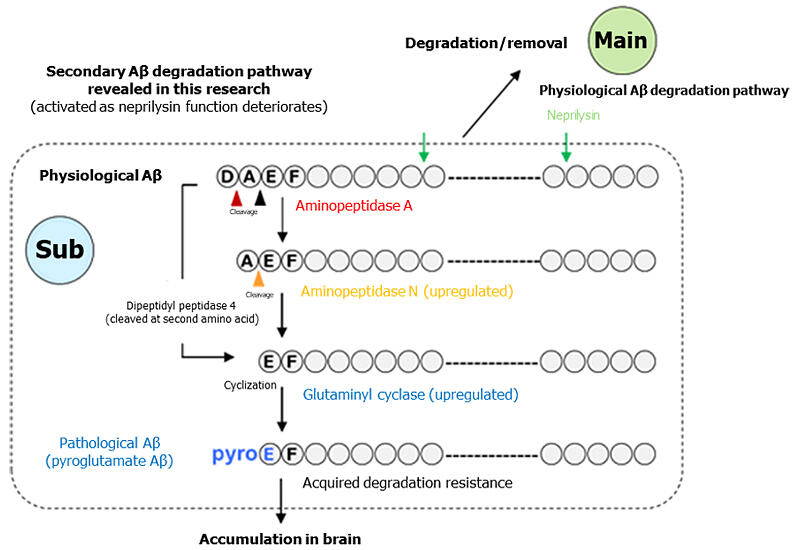
Most of the Aβ molecules that accumulate and form amyloid plaques in the AD brain are in a pathological Aβ form called pyroglutamate-Aβ. This form has a 250-fold higher self-aggregating capacity and structurally differs from physiological Aβ produced in the normal brain. Physiological Aβ is degraded and removed by neprilysin, but AD patients have been shown to have an extremely reduced ability to produce neprilysin in the brain. Aβ in the brain is repeatedly produced and degraded and accumulates over a long period of time until the onset of AD, during which the amino-terminal structure is known to undergo various modifications.
Until now, due to the high aggregation propensity of pyroglutamate-Aβ, accumulated pyroglutamate-Aβ has not been analyzed quantitatively. The mechanism by which physiological Aβ is transformed into pathological Aβ also remained unknown.
In this study, the research group chemically synthesized various amino-terminal variants of Aβ labeled with radioisotopes to distinguish the Aβ forms present at certain levels in the brain and injected them into the hippocampus of rats to analyze their susceptibility to degradation. As a result, physiological Aβ and Aβ variants in which only amino-terminal residues were removed were rapidly degraded. However, pyroglutamate-Aβ, in which the amino terminus was cyclized after amino-terminal residues were removed, was degraded extremely slowly.
Quantitative analysis of the abundance of these various Aβ variants in brain autopsies of AD patients using specific antibodies revealed that pyroglutamate-Aβ accounted for at least 40% of the total amount of the Aβ peptides, whereas physiological Aβ did not exceed 5%. The research group genetically engineered the conventional AD model and next-generation AD model mice to become deficient in the neprilysin function. These neprilysin-deficient AD model mice were compared with AD model mouse brains with functional neprilysin for analysis. The analysis demonstrates that amounts of brain Aβ with the same amino-terminal structure as physiological Aβ were higher in the former model mice than in the latter model mice until the age of 12 months, after which the difference became smaller. Pyroglutamate-Aβ accumulated in an age-dependent manner in the model mouse brains without neprilysin function, reaching 2.5-fold at 24 months of age. They also devised a pretreatment method for the mass spectrometric detection of pyroglutamate-Aβ, which was impossible before.
Using Pittsburgh compound B (PiB) as an imaging probe, they successfully captured increases in the brain pyroglutamate-Aβ level through positron emission tomography (PET). Pyroglutamate-Aβ has been shown to undergo cleavage of the two amino-terminal amino acids from physiological Aβ, followed by cyclization of glutamic acid at position 3.
Previously, Saido showed that exopeptidases, which include aminopeptidases and dipeptidyl peptidases, are enzymes cleaving amino acids from the amino terminus of peptides, were involved in the production of pyroglutamate-Aβ. Therefore, they examined the possibility that exopeptidases compensate for deficient neprilysin function in Aβ degradation.
With a focus on aminopeptidases, dipeptidyl peptidases, and glutaminyl cyclases as possible enzymes involved in Aβ degradation, they tested reactions of these enzymes with Aβ and found that all of these enzymes had sufficient cleavage activity. The research team also confirmed that brain expression levels of aminopeptidases and dipeptidyl peptidases increased in an age-dependent manner in neprilysin-deficient AD model mice. These enzymes have cleavage activity, but adding the degradation-resistant "accessory" to Aβ by glutaminyl cyclases during the degradation process was found to preclude further degradation, leading to progressive accumulation of pathological Aβ.
This study demonstrated the therapeutic potential of inhibitors of aminopeptidases, dipeptidyl peptidases, and glutaminyl cyclases. These inhibitors work before pathological pyroglutamate-Aβ formation is completed, unlike donanemab, which targets and removes pyroglutamate-Aβ already accumulated in the brain.
Saido said, "Currently, two antibody drugs for AD, lecanemab and donanemab, have been approved by the U.S. FDA, the Ministry of Health, Labour and Welfare of Japan, and others. The latter is an antibody that was discovered by myself and Ihara (the University of Tokyo) in 1995 and recognizes pyroglutamate-Aβ, while lecanemab is an antibody that was produced by Lannfelt et al. (Uppsala University) and recognizes soluble Aβ oligomers. Because these antibodies recognize different epitopes (a site recognized by an antibody at the molecular level), additive effects may be seen when used together. However, caution should be exercised in adverse effects such as intracerebral hemorrhage."
Journal Information
Publication: Life Science Alliance
Title: Metabolic resistance of Aβ3pE-42, a target epitope of the anti-Alzheimer therapeutic antibody, donanemab
DOI: 10.26508/lsa.202402650
This article has been translated by JST with permission from The Science News Ltd. (https://sci-news.co.jp/). Unauthorized reproduction of the article and photographs is prohibited.